|
GENODERMATOSES
|
|
|
|
General considerations Genodermatoses is referred to a group of diseases which are due to genetically determined disorders. A congenital abnormality which is present at or before birth, is not necessarily genetically determined. A congenital defect: may be as a result of an infection from the mother or an abnormality in the development, which is not genetically related. Not all inherited disorders are congenital; they may not become apparent until late childhood or even old age. The bodily manifestations of the genotype are referred to as the phenotype. Manifestations tend to appear when the organ or tissue concerned first reaches its full functional development. Some common skin disorders such as atopic eczema and psoriasis are manifestations of abnormal constitutional states of genetic origin. Genetic Principles An idea about genetic theory is essential for the understanding the terms or syndromes to be discussed. Hereditary characteristics are transmitted from one generation to the next by chromosomes, composed of double helix strands of deoxyribonucleic acid (DNA). A gene is a sequence of bases in DNA that codes for one polypeptide. The precise position of the gene on the chromosome is known as its locus. In females the 46 chromosomes are present in homologous pairs and thus there are two copies of every gene, one maternal and the other paternal in origin. It is the same in males except for the difference in the sex chromosome pair X and Y. Alternative genes at a single locus are called alleles. An individual with two different alleles at a particular locus is heterozygous for that gene; where both alleles are identical; the individual is homozygous for that gene. Different Types of Genes Dominant gene: capable of exerting its full effect when it is present on only one member of the chromosome pair (heterozygous state). Recessive gene: the gene is present at both corresponding loci (homozygous state) before it can exert its full effect. Those genes borne on chromosomes other than the sex chromosomes (X and Y) are known as autosomal. Characters controlled by genes borne on the X or Y-chromosomes are sex-linked. The Y chromosome is much smaller than the X chromosome. X-linked inheritance is the only one of significance in clinical practice. The great majority, perhaps all, sex-linked genes are exclusive to the X chromosome. Mutations Normally, replication of DNA is completely accurate, but errors or mutations can occur at random. Mutations may occur as a result of point substitution of a single nucleotide base or by insertion or deletion of one or two base pairs. If a mutation occurs in a somatic cell, (somatic mutation), only the descendants of that cell are affected and there will be no transmission of the abnormality to further generations. Only mutations occurring in the gametes or their precursors can be transmitted to offspring. Genetic Linkage and Diseases Association Genes residing on the same chromosome remain linked in transmission so long as the chromosome remains intact, but during reduction division or meiosis such linkages may be disrupted if crossing-over occurs. The closer two genes are situated on a chromosome, the less likely they are to be separated by crossing-over and the more likely they are to be inherited together. Two such gene loci are said to be linked and it is possible to demonstrate genetic linkage in a family using appropriate genetic markers, if different alleles are present at each of the two loci. When two alleles occur together, the case is in linkage disequilibrium, which may result from recent mutation or of a particular combination. Genetic Counselling Genetic counseling depends upon the recurrence risk to parents of having an affected child. Both parents should be investigated thoroughly. If the abnormal character is determined by an autosomal dominant gene and one parent is affected, 50% of the offspring will be affected. Some parents fail to understand that the risk remains constant for every pregnancy, and that an affected first child does not guarantee a normal second child. If the parents of an affected child have no manifestations of genetic abnormality, the recurrence risk is likely to be small, as the child would have the genetic disorder as a fresh mutation. Autosomal recessive disorders are homozygous for the mutant gene. The recurrence risk for the carrier parents is 1 in 4, but offspring risk for those who are affected is small. Most recessive conditions are rare and it is unlikely that the affected person will marry another carrier. Histocompatibility Antigens (HLA) HLA antigens are glycoproteins on the cell surface of most nucleated human cells. These differ from person to person and uniquely fingerprint to each person‘s cells. These fingerprints allow a person‘s immune system to be recognized if a given cell is its own. The importance of the HLA antigens is of prime importance in matching donors and recipients in the transplantation of human tissues. The HLA region is located on the short arm of chromosome 6, referred to as the major histocompatibility complex (MHC). A person inherits HLA antigens as a set; one set (haplotype) from each parent. There are at least 4 or 5 genetic loci that produce HLA antigens termed A, B, C, D and DR and their gene products are called HLA-A, -B, -C, -D and -DR antigens. Each locus has multiple allelic determinants (polymorphism). Each allele at each locus controls an antigen that is identified by a number placed after the letter of that series, e.g. HLA-A1, HLA-B5. The association of an HLA antigen with a given disease means that there is a higher incidence of that antigen in a group of patients with the disease than in a group of people without the disease. Molecular mimicry An infective agent may have a similar configuration to the HLA antigen, so that the agent is then not attacked by the body‘s defense system. Alternatively, the agent might differ only slightly from the HLA antigen, so those antibodies are produced which attack both the infective agent and the cells, which contain the HLA antigen, thus inducing autoimmune damage. Genetic linkage The HLA antigen may be close to another gene on the same chromosome which produces a disease, either directly (e.g. due to an enzyme deficiency) or indirectly due to an effect on the immune response, which may be either abnormally enhanced, leading to autoimmunity or abnormally decreased leading to infection. Receptor effects Many chemicals, including drugs and toxins, bind to the cell surface before they are taken into the cytoplasm. Since HLA antigens are present on the cell surface, they could modify the binding of these potentially toxic substances. The association between HLA and a particular disease is not absolute. Chromosomal Disorders Chromosomal disorders may be due to abnormalities of chromosome number or structure. They may involve autosomes or sex chromosomes. Approximately 7.5% of all conceptions have a chromosomal disorder, but most of these are spontaneously aborted. Chromosomal abnormalities generally cause multiple congenital malformations. Children with more than one physical abnormality, should undergo chromosomal analysis as part of their investigation. Chromosomal disorders are incurable but can be reliably detected by prenatal diagnostic techniques. Amniocentesis or chorionic villus sampling should be offered to women whose pregnancies are at increased risk, namely, women in their mid-thirties or older and couples with an affected child. Skin Manifestations of Chromosomal Disorders Cutaneous manifestations of different autosomal defect syndromes are discussed briefly below:
DOWN‘S
SYNDROME It is the most common autosomal abnormality, characterized by bluish black macules of various sizes and shapes occurring on the sacral region of the newborn, mostly in orientals. Mongolian spots usually disappear during childhood. Down‘s syndrome accounts for about one-third of all moderate and severe mental handicap in children of school age. Etiology Most cases result from trisome of chromosome 21 in which the extra chromosome is derived by non-dysfunction at meiosis usually from the mother. The affected child has the normal number of 46 chromosomes but one of the clinically normal parents carries a translocation of part of chromosome 21. Clinical Manifestations General manifestations The facial appearance is diagnostic and is characterized by the following: Small head, flat face with small ears . Short nose. Mongoloid eyes with slanting pulpebral fissures and epicanthic folds. Thickened eyelids with short and sparse eyelashes. Hypoplastic iris with hypopigmented spots (Brush field spots). The limbs are stumpy and the joint ligaments are lax. The fingers are short and cone-shaped and are sometimes webbed. The little finger is often curved. Skin manifestations - At birth
the skin is normal. Generalized xerosis at the age of 15. Patchy lichenifecation showing dry skin surface. A chronic follicular papular eruption of the presternal and interscapular region. Skin infections, angular cheilitis, chronic blepharitis and a purulent nasal discharge are common. Tinea pedis: The cheeks are often red. Peripheral circulation is poor, acrocyanosis is frequent and livedo reticularis is often conspicuous throughout the year, on the thighs, buttocks and trunk. Dermatoglyphic features include a single flexion crease on the fifth finger, and an increased incidence of ulnar loops on the fingers. Mucous membrane manifestations Fissuring and thickening of the lips. The tongue is scrotal in almost all cases. Elastosis perforans and syringomata occur more often than in normal subjects. Hair manifestations The hair may be normal, but is often fine and may be hypopigmented. Alopecia areata. Teeth manifestations The teeth are hypoplastic and late to erupt. Other manifestations
When serious malformations are present, death during infancy is common.
This is the second most common multiple malformation syndrome. It occurs in about 1 per 3000 live births, 95% of affected fetuses abort spontaneously. Parental dysfunction at either the first or second meiotic division results in the extra copy of chromosome 18. Rarely, a parental translocation is responsible. Occasionally, mosaicism is seen with a milder phenotype. General Manifestations The syndrome comprises severe mental deficiency with a characteristic skull shape and a small chin. Other general features are: Prominent occiput. Low-set malformed ears. Clenched hands with overlapping index and fifth fingers. Single palmer crease Rocker-bottom feet and short sternum. Malformations of the heart, kidneys and other organs are frequent. Cutaneous features: these include cutis laxa of the neck, hypertrichosis of the forehead and back. Capillary hemangiomas. Fingerprints show a distinctive low-arch dermal ridge pattern. Thirty per cent die within a month. Only 10% survive beyond the first year and these infants show profound developmental delay.
The characteristic features of the syndrome are: General Manifestations Mental retardation. Sloping forehead . Eye defects, including microphthalmia or anophthalmia. Cleft palate, cleft lip and low-set ears. Rocker-bottom feet. Cardiac defects and a variety of other visceral abnormalities. Survival for more than 6 months is unusual. Cutaneous features Capillary hemangiomas, especially that of the forehead, hyperconvex nails and localized defects of the scalp. Cutis laxa of the neck has also been reported. The palm prints shows distal palmer axial triradii.
TURNER‘S
SYNDROME The frequency of Turner‘s syndrome is 1 per 2500 of female births. In some 80% of cases there are 45 chromosomes with an XO sex chromosome complement. Such cases are chromatin-negative buccal smears. It is assumed that the missing chromosome was lost before or at fertilization. The incidence 45X is increased in the offspring of teenage mothers. Most of the remaining 20% of cases are chromatin-positive. Some have 46 chromosomes but with part deletion of one X chromosome. Other cases have shown mosaicism of various types, XX/XO or XXX/XO. Clinical Manifestations Webbed neck Low posterior hairline margin. Alopecia of the frontal area of the scalp. Triangular mouth. Short stature. Increased carrying angle of the elbow. Koilonychia. Mental retardation.
This syndrome occurs in both sexes as pheno-type and resembles Turner‘s syndrome. This is considered by others as “ the male type Turner‘s syndrome “ but the karyotype is normal (46XY or 46XX). Most cases are sporadic, but autosomal dominant inheritance has been reported. Noonan‘s syndrome is most common in males. Clinical Manifestations The features show characteristic association of hypertelorism, blepharoptosis, epicanthic folds and a small chin. Short stature and have a broad, short neck that may be webbed. Skeletal defects are frequent. Low hairline in the back and the hair is coarse, light colored and curly with a low posterior hairline Downy hypertrichosis may occur on the cheeks or shoulders. Pubic hair is scanty in the male and beard growth is poor. Heart defects and pulmonary stenosis are often present. Intelligence may be normal but some degree of mental retardation is usual. In 70% of males the testes are undescended. Lymphoedema of the feet and legs is common and more severe than in Turner‘s syndrome. Low -set ears. Cubitus vulgaris. Differential Diagnosis Turner‘s syndrome Absence of coaractation of the aorta and its prevalence in males. In contrast to Turner‘s syndrome, short stature and infertility are not constant features. Ulerythema ophryogenes presents with cutaneous manifestations as that of Noonan‘s syndrome.
The diagnosis of Noonan‘s syndrome must be suspected in all patients labeled as Turner‘s syndrome if they are of normal height, mentally retarded, have a cardiac-valve defect or with normal gonadal function.
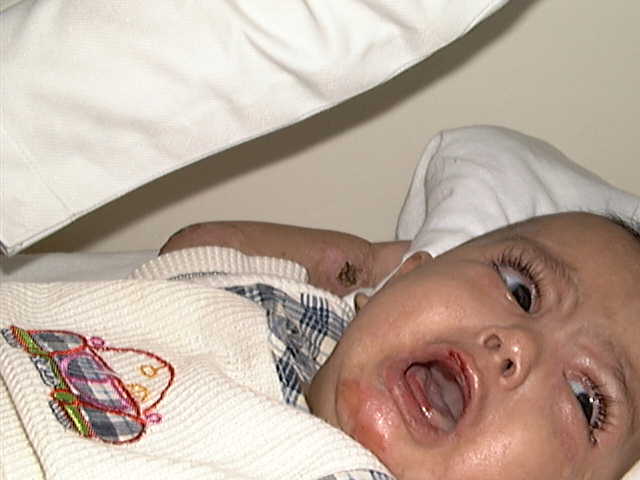
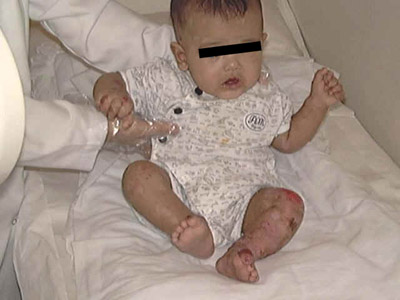
Klinefelter syndrome is a problem of male sex differentiation; it is frequently an XXY sex chromosome pattern. General Manifestations Klinfilter‘s syndrome affects male births and is characterized by: Hypogonadism Gynecomastia
Fig. 252a. KlineFilter's syndrome
Fig. 252b. KlineFilter's syndrome &Icthyosis
Eunuchoidism Some patients are tall and obese. Small or absent testicles. Elevated serum gonadotrophin. Skin manifestations Minimal but there may be a low frontal hair line, sparse body hair and absent or very few hair on the pubic, axillary and beard areas. Shortening of the fifth digit of both hands. Vascular manifestations: acrocyanosis, vascular angiomas, peripheral vascular disease and stasis dermatitis. Psychiatric disorders occur in about one third of the patients. Associated features: Osteoporosis, leg ulcers, obesity, psychiatric disorders and taurodontism (vertical enlargement of the molar pulp chamber). Diagnosis The association of gynecomastia with small testes and otherwise apparently normal genitalia should suggest the diagnosis, which is supported by finding an increased urinary excretion of gonadotrophin. The diagnosis is confirmed by chromosome studies. Treatment Testosterone replacement therapy will improve secondary sexual characteristics, but not the infertility.
NEUROFIBROMATOSIS This syndrome is a congenital dysplasia characterized by cutaneous, nervous system, muscles and bones manifestations. Clinical Manifestations Skin manifestations Characteristic skin manifestations include café au lait macules. Café au lait macules are flat and round with several dark brown spots. When there is six or more of these macules of a size of at least 1.5-cm in diameter, the diagnosis of neurofibromatosis is established. Café au lait spots are the first features of the disease to appear in all children. Bronzing of the skin pigmented hairy nevi; axillary freckling and sacral hypertrichosis are other manifestations of the disease.
Multiple neurofibromas occur along peripheral nerves which are soft and cystic, pedunculated, most numerous on the trunk and limbs. Hundreds may be present, ranging from a few millimeters to several centimeters in diameter. The other type of the disease is characterized by the occurrence of acoustic neuromas, usually bilateral, as well as meningiomas and other tumors of the nervous system. Mucous membrane manifestations Oral lesions are present in 5-10% of cases, as papillomatous tumors of palate, buccal mucous membrane, tongue and lips, or as macroglossia, which is usually unilateral. Ocular manifestations Lisch nodules (melanocytic pigmented iris hamartomas) appear as dome-shaped lesions found superficially around the iris on slit-lamp examination. Pruritus may be a symptom of neurofibromatosis. The presence of large numbers of mast cells in the skin in this condition, and the response of the itching to antihistamines suggest that histamine is the cause of pruritus. Elephantiasis neurofibromatosa is a similar diffuse neurofibromatosis of nerve trunks associated with overgrowth of the subcutaneous tissue and of the skin, which is wrinkled and pendulous and may produce gross disfigurement. Internal manifestations Neurofibromas may also involve the viscera and blood vessels. Bone changes Kyphoscoliosis occurs in 2% of cases, and the early onset leading to cardio-respiratory disease, unless aggressive surgery is performed. Osteomalacia when present is the result of a congenital defect of the renal tubules. Pseudoarthrosis involving the tibia or radius. Short stature and macrocephaly. Intellectual handicap occurs in one third of cases and physical development may be impaired. Speech abnormalities. Hypertelorism and headaches are also common. Endocrine disturbances These include: Precocious puberty, Acromegaly, Addison‘s disease, hyperparathyroidism, gynecomastia and phaeochromocytoma of the adrenal. Reno-vascular hypertension may occur in children. Involvement of the lower urinary tract may give rise to urinary symptoms. Gastro-intestinal manifestations Constipation occurs due to dysfunction of the colonic musculature. Gastrointestinal lesions may also cause recurrent hemorrhage or obstruction. Neurological manifestations The most common solitary intracranial tumor is an optic nerve glioma. Astrocytomas and Schwannomas also occur. Tumors may arise in peripheral nerves and within the spinal cord. Intracranial tumors may cause epilepsy, but fits may occur in the absence of any demonstrable focal lesion. Malignancy: sarcomatous changes may accompany the disease. Malignant change may occur simultaneously in several lesions. Enlargement or pain should suggest the possibility of malignant change, but rapid enlargement may follow hemorrhage. Other malignant diseases associated with neurofibromatosis include Wilms‘ tumor, rhabdomyosarcoma, leukemia and retinoblastoma. Course and Prognosis The course of the disease varies considerably. Characteristically café au lait spots are present either at birth or, more commonly, develop in early childhood. Cutaneous neurofibromas appear usually during childhood and increase rapidly in number at puberty. However, lesions may be present at birth and become progressively more extensive. Although early onset and rapid progression before puberty usually indicate a poor prognosis, minimal cutaneous involvement in the young child does not necessarily imply a favorable course, although many cases remain limited. Extensive involvement of the urinary or gastrointestinal tract or of the central nervous system carries a poor prognosis. Very rarely, the disease may be so extensive at birth to endanger life. Pregnancy induces rapid progression of existing lesions and the development of new ones besides hypertension. Diagnosis Cutaneous neurofibromas are clinically and histologically distinctive. Café au lait spots, usually are the earliest manifestations in children. These are present in 10-20% of normal individuals and in about 35% of patients with Albright syndrome. If six or more café au lait lesions are present, the possibility of neurofibromatosis is high, and if these are associated with axillary freckling the diagnosis is almost certain. Epilepsy should be thoroughly investigated. Prolonged follow-up with routine checks every 6-12 months is advisable. Genetic counseling is important. First-degree relatives (e.g. siblings and offspring) who have no stigmata of the disease are unlikely to carry the gene and the risk for their offspring is minimal. All patients should be thoroughly investigated, to include an IQ assessment, electroencephalography, audiography, slit-lamp ocular examination, radiological skeletal survey, cranial CT scan and 24-h Urinary catecholamine assays. Treatment Treatment is symptomatic. The more disfiguring lesions can be excised if not too diffuse. Surgery is also indicated when an increase in size and pain suggests possible malignant change. We used CO2 laser for excision of cystic lesions of neurofibromas in a young female using topical Emla cream as local anaesthetics. The patient tolerated the procedure well and was satisfied with the result.
This condition was originally considered to be part of the spectrum of Von Recklinghausen‘s disease, but it is now recognized as a separate entity, because of its distinct genetic basis and natural history. Café au lait spots and cutaneous neurofibromas may be seen, but are usually few in numbers and much less common than in NF-1.
TUBEROUS
SCLEROSIS Tuberous sclerosis is a complex genetic disorder transmitted as an autosomal dominant gene. The syndrome is characterized by triad: epilepsy, mental deficiency and adenoma sebaceous. The characteristic skin lesions are angiofibromas in the form of pin-head - sized yellowish red, translucent, discrete, waxy papules situated symmetrically on the face. Although tuberous sclerosis and neurofibromatosis have certain features in common and may coexist, they are genetically distinct. Clinical Manifestations The characteristic features of the syndrome are: Skin manifestations Skin lesions are found in 60-70% of cases. Lesions of four types are pathogonomonic.
Other cutaneous manifestations include: Firm, fibromatous plaques, especially on the forehead and scalp. Soft, pedunculated fibromas may appear around the neck, axillae and poliosis. Mucous membrane manifestations Oral papillomatosis on the gingiva and on other parts of the mouth and nose. Fibromatous tumors are occasionally present on the gums, palate and are rarely found on the tongue, larynx or pharynx. Teeth manifestations Small pits commonly occur in the tooth enamel in adult patients, and these pits, though less obvious in the deciduous teeth, have been used as an early diagnostic sign in children with tuberous sclerosis. Nail manifestations Sublingual fibromas and Koenen‘s periungual tumors appear as small degitate protruding asymptomatic tumors. Neurological manifestations Epilepsy: may appear early in infancy. Mental deficiency is usually seen early during infancy. Psychotic symptoms, including schizophrenia. Bone changes: osteoporosis of the long bones and skull with pseudocysts mainly in the phalanges. Eye changes: retinal tumors Renal tumors in the form of hamartomas. Cardiac tumors as rhabdomyoma. Pulmonary changes These changes are rare and seldom cause symptoms but if they are extensive, they may cause increasing dyspnea and recurrent spontaneous pneumothorax. Gastrointestinal tumors These are usually hamartomatous colonic polyps. Colonoscopy should be considered in the investigation of patients with tuberous sclerosis. Endocrine manifestations Endocrine and other metabolic disturbances may be present, most frequently reported are pituitary-adrenal dysfunction, thyroid disorders and premature puberty. Primary localized gigantism and diffuse cutaneous reticulohistiocytosis. Radiological Findings Skull Calcification is seen on plain skull X-ray in about 50% of patients. These are not usually apparent until later childhood or adult life. The typical CT scan appearance of tuberous sclerosis consists of calcified periventricular nodules that project into the lateral ventricles. MRI imaging is more sensitive in the detection of parenchymal lesions. The periventricular lesions may not be seen initially and can progress to calcified lesions with time. Hands and feet: Cyst-like lesions of the phalanges and irregular thickening of the cortex of metatarsals, vertebrae, pelvis or long bones are not uncommon. Lungs There may be irregular reticulation of the lung-fields, not radiologically distinguishable from other types of interstitial fibrosis. Treatment No specific treatment is available. Symptomatic treatment of the affected organs.
Epidermolysis bullosa (EB) is a rare chronic skin diseases characterized by formation of bullae elicited by friction or trauma to the skin due to separation at the dermo-epidermal junction. Epidermolysis bullosa is either a genetic dominantly transmitted disease or acquired.
TYPES OF EPIDERMOLYSIS BULLOSA Epidermolysis bullosa simplex Dystrophic dominant Dystrophic recessive
This is a dominantly recessive type, characterized by formation of bullae mainly on the palms and soles elicited by friction or trauma. The lesions become apparent when the child starts to crawl or walk, where the bullae appear after minor trauma or friction. Healing or rupture of the bullae takes long time without scarring. Hair, nails or buccal mucosa are not affected. Treatment Preventive measures are the main line of treatment, which includes minimizing friction or trauma by using loose or open shoes, and avoiding trauma. Symptomatic Treatment. Treatment of secondary infection. Corticosteroid is helpful in severe cases. Topical potent fluorinated steroids cream for skin lesions. Care should be considered in such type of treatment because of possibility of absorption of the concentrated steroids, hence, the skin is abraded that enhances more absorption leading to unwanted side effects. Some authors reported the success of oral sodium citrate 2gms three times daily.
Fig. 254 Epidermolysis bullosa
The lesions are precipitated by friction and trauma where bullae appear due to subepidermal splitting. The lesion has a chronic course healing by scarring. Mucous membranes and hair are not affected . Nickolysky‘s sign is usually positive and the bullae are flaccid where the fluid in the bulla can be moved few centimeters.
|
||||||||||||||||||||||||







| Contents | << Previous Chapter | Next Chapter >> | Search |